


Infer, filter and enhance topological signals in single-cell data using spectral template matching
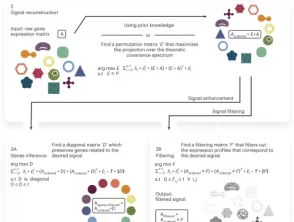
Single-cell RNA sequencing is a powerful technology that allows researchers to analyze gene expression in individual cells, providing insights into cellular processes and functions. However, analyzing this data can be challenging, as cells can simultaneously encode multiple, potentially cross-interfering, biological signals. A new computational method, scPrisma, was developed to address this challenge. scPrisma has the ability to uncover cellular spatiotemporal context and has the potential to drive further insights into cellular processes and functions, ultimately advancing our understanding of biology. You can check out the published article at: https://www.nature.com/articles/s41587-023-01663-5
Goal
Infer, filter and enhance topological signals in single-cell data using spectral template matching
Researchers
- Jonathan Karin
- Yonathan Bornfeld
- Mor Nitzan
What we did
We apply scPrisma to the analysis of the cell cycle in HeLa cells, circadian rhythm and spatial zonation in liver lobules, diurnal cycle in Chlamydomonas and circadian rhythm in the suprachiasmatic nucleus in the brain. scPrisma can be used to distinguish mixed cellular populations by specific characteristics such as cell type and uncover regulatory networks and cell–cell interactions specific to predefined biological signals, such as the circadian rhythm. We show scPrisma’s flexibility in incorporating prior knowledge, inference of topologically informative genes and generalization to additional diverse templates and systems. scPrisma can be used as a stand-alone workflow for signal analysis and as a prior step for downstream single-cell analysis.
Results
https://www.nature.com/articles/s41587-023-01663-5
The complexity of social complexity
Developing quantitative multidimensional approaches for studies on social complexity
The evolution of complex behaviours is a major challenge in evolutionary and behavioural biology. The current “Omics” revolution in biology opens new opportunities to study this problem in molecular terms, but this requires the development of new statistical and data analyses tools. We propose to start addressing this challenge by focusing of the evolution of social complexity in insects. Research of social insects such as ants, termites, bees and wasps has provided excellent model systems for developing hypotheses and theories on the evolution of social complexity. These include seminal contributions such as the development of kin selection theory, multi-level selection theory, and the influential idea that the evolution of complexity has progressed through a series of major transitions. Currently, the evolution of social complexity in insects relies on qualitative classifications. We argue that this approach suffers from several significant limitations. These include, lumping together species showing a broad range of social complexity, and falsely implying that social evolution always progresses along a single linear stepwise trajectory that can be deduced from comparing extant species (“rungs on a ladder”). We recently showed that a single species can have both higher and lower levels of social complexity compared to other taxa, depending on the social trait measured. This study proposes a new approach which is based on measuring the complexity of individual key social traits.
Goal
Developing statistical and data analyses tools for studying social complexity within an appropriate phylogenetic framework.
Researchers
What we did
In progress
Results
Forthcoming
Integrating large-scale datasets into a metamodel of multiscale communication networks underlying Alzhimer’s disease progression
Alzheimer’s disease (AD) is a progressive neurodegenerative disease of old age and the most common cause of dementia. Despite great advances made in understanding the pathogenic features leading to AD, we still only partially understand its cause, and currently have no effective treatments and prevention strategies. While AD research has been largely focused on the damage to neuronal cells, accumulating evidence suggests that multiple non-neuronal cell types in the brain are directly involved in the degeneration process. Cellular communication between different types of brain cells are predicted to have a major contribution to AD progression, yet much remains unknown regarding cellular communication networks spanning multiple cell types.
We are interested in expanding the research focus from single cell types to profiling entire cellular environments, aiming to build a model of the cellular cascade leading to AD, i.e. the crosstalk between cell types and the consequent changes in their internal cell states that drives the disease. We are relying on our combined expertise in: applying cutting edge genomics and imaging technologies in the brain to generate large scale datasets with single cell resolution; in machine learning, image analysis, and graph theory approaches to tackle the data analysis challenges; and in integrative modeling of dynamic biological systems across scales using Bayesian modeling approaches.
Goal
To create a comprehensive multi-resolution model that describes the cascade of cellular communications during Alzheimer’s disease (AD) progression
Researchers
What we did
In progress
Results
Forthcoming
Preimplantation embryo quality
Goal
More than one out of every 25 babies in Israel is a test-tube baby, but there is still no accurate means of evaluating embryo quality prior to implantation. Thus, multiple embryos are implanted leading to reasonable pregnancy rates but also increasing the rate of multiples. The aim of this project is to predict which pre-implantation embryos have the best chance of leading to successful pregnancy.
Researchers
What we did
We collected a dataset based on over 70,000 embryos (Nanshe Database) and conduct the first large-scale statistical analysis of preimplantation development.
Results
We achieve superior classification of embryo blastulation and implantation.